
2024年2月29日,西安交通大学李磊团队在Nature Communications 发表题为“MYC induces CDK4/6 inhibitors resistance by promoting pRB1 degradation”的研究论文,本研究证明了在膀胱,前列腺和乳腺癌细胞中MYC扩增导致CDK4/6i耐药。在机制上,MYC与E3泛素连接酶 KLHL42 的启动子结合并增强其转录,通过诱导磷酸化和全部pRB1泛素化和降解导致RB1缺乏。
该研究鉴定了一种降解MYC的化合物 A80.2HCl,它在纳摩尔浓度下诱导MYC降解,恢复pRB1蛋白水平并重新建立MYC 高表达癌细胞对CDK4/6i的敏感性。CDK4/6i和A80.2HCl联合应用可显著抑制体内肿瘤生长。总而言之,这些结果揭示了MYC诱导的CDK4/6i耐药性的分子机制,并表明利用MYC降解分子A80.2HCl来增强CDK4/6i的治疗效果。
癌症通常被定义为由控制细胞周期的分子事件严格调控的细胞增殖疾病。细胞周期蛋白依赖性激酶CDK4和CDK6 (CDK4/6)调控细胞周期早期G1期的进展,因此它们对癌症治疗显示出良好的易感性。靶向CDK4/6的药物抑制剂已显示出对多种实体瘤的深远效果,并已被批准用于治疗激素受体(HR)阳性、人表皮生长因子受体2(HER2)阴性的晚期或转移性乳腺癌。然而,其他癌症类型的患者可能无法从CDK4/6i中获益。CDK4/6i的内在耐药机制及其在其他癌症类型中的应用值得进一步研究。
目前已经确定了对CDK4/6抑制产生耐药性的许多推测机制,在这些机制中,正常RB1功能的丧失是对CDK4/6i耐药的细胞中最常观察到的变化,这主要是因为pRB1作为主要CDK4/6底物发挥了典型作用。pRB1通过结合和隔离E2F转录因子来负调节细胞周期进程,从而阻止进入细胞周期的S期。pRB1通过CDK4/6磷酸化导致E2F转录因子的释放,最终驱动细胞周期进程。RB1缺失是许多癌症中最基本的事件之一。虽然RB1的基因缺失已被广泛研究,但pRB1蛋白在翻译后水平失活仍然知之甚少。
MYC是研究最广泛的癌蛋白之一,可调节许多细胞过程,并有助于几种不同癌症类型的肿瘤发生和治疗耐药性。MYC家族包括广泛表达的MYC、分布较不广泛的MYCN和广泛研究的MYCL家族成员。MYC通过与多种因子和复合物的相互作用,作为转录的通用扩增器发挥作用。MYC基因在多达70%的人类癌症中过表达,这使得MYC成为癌症治疗的一个有吸引力的理论靶点。在多种肿瘤模型中沉默MYC导致与肿瘤微环境重塑相关的肿瘤消退。尽管已经进行了多项努力,但用临床级小分子靶向MYC仍然是一个很大的挑战,特别是在蛋白质水平上。
在本研究中,发现高MYC表达通过直接激活E3泛素连接酶KLHL42的转录来驱动对 CDK4/6i的耐药性,从而促进pRB1泛素化和降解,从而导致pRB1蛋白缺乏。此外,研究表征了一种可降解MYC的分子A80.2HCl,该分子在纳摩尔水平应用时可消除MYC,挽救pRB1蛋白活性,并降低MYC依赖性CDK4/6i耐药性。此外,CDK4/6i和MYC降解分子A80.2HCl的组合在体内外均显示出对肿瘤细胞的协同杀伤作用。
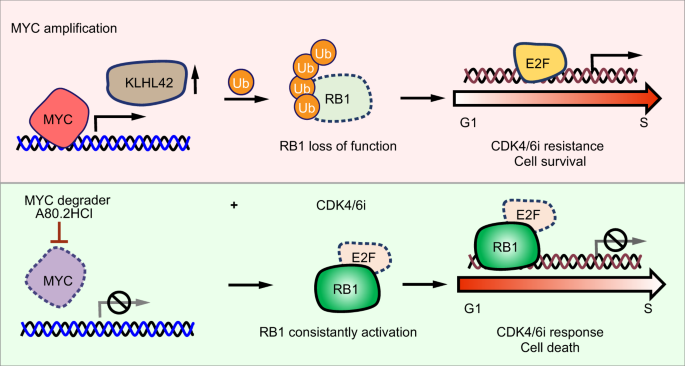
工作模型示意图(图源自Nature Communications )
总之,该研究揭示了MYC通过破坏pRB1蛋白稳定性在驱动CDK4/6i抗性方面以前未被认识到的作用。高MYC表达激活E3连接酶KLHL42,该连接酶负责pRB1泛素化和降解,随后使癌细胞对CDK4/6i耐药。此外,研究发现了一种 MYC 降解分子A80.2HCl,可有效降低 MYC 蛋白水平并克服CDK4/6i耐药,阐明了CDK4/6i在癌症治疗中扩大使用的策略。
参考消息:
https://doi.org/10.1038/s41467-024-45796-w