

介绍
结构变异(structural variants,SV)包括基因组重排的多种形式,例如拷贝数变异(copy number variation, CNV)、移动元件插入(mobile element insertions, MEIs)、倒位、易位等。典型的人类基因组中包含7000-25000个多态SV,大多数为双等位基因CNV或MEIs。大多数SV对基因功能影响很小或没有影响,但也有SV可以成为先天性疾病的致病变异。
通常使用染色体微阵列(chromosomal microarrays, CMA)来检测大型SV(通常>20kb),其特点是成本低,但是分辨率也较低。外显子检测(exome-sequencing, ES)在临床上被应用于检测单核苷酸变异和小插入/缺失,但是CMA仍是检测SV的主流,其中有几个原因。首先,测序的成本,计算能力和信息复杂性是一个障碍;其次,当前的基于ES的SV检测方法侧重于通过覆盖度差异确定拷贝数变异。因此,ES探测CNV的分辨率大小与CMA相似,一般限制在>10kb。尽管现在ES作为SV的检测方法比CMA更有潜力,在临床上仍进展缓慢。
因此大于50bp小于10kb的变异难以检测到,作者将之称为“隐藏”的变异。为了检测这些变异,作者开发了InDelible工具,通过检测ES数据中代表SV断裂点的拆分reads寻找SV。作者将重点放在拆分reads上,因为不同SV的共同特征就是会形成独特的连接序列。随后作者使用InDelible分析了13438个严重发育障碍(developmental disorders,DD)先证者的ES数据。
InDelible分析SV需要多个步骤,总结一下就是识别拆分的reads,将它们集中到同一基因组位置的簇中,过滤这些簇来去除测序原因造成的伪影并保留可能存在的变异,然后组合拆分reads的未对比部分并定位到基因组中。InDelible还计算了每个拆分reads簇在人群中的频率,以便根据次等位基因频率(MAF)筛选变异。
与GATK和Menta的对比
使用三种方法对来自the Genome in aBottle Consortium同一个样本的ES数据进行分析,并以此样本在the Genome in a Bottle Consortium数据库中的变异数据集作为金标准。这个数据集集合了短读长测序,连锁分析和长度长测序的变异结果来检测每种算法的召回率和特异性。
使用ES数据,InDelible在21bp和10kb之间变异的召回率相仿或优于GATK和Menta。假阳性率略高与GATK,这是因为InDelible被设计为最大灵敏度,以检测临床变异。
[注:召回率(recall)= TP/(TP+FN),代表查全率]

InDelible在DD患者队列中的应用
InDelible设计的一个主要目标是识别先证者的新发变异。因此变异的过滤条件为:1,人群频率低;2,未受累的父母不携带(如果有父母数据);3,变异的断裂点在已知疾病相关基因的编码序列内。
作者使用InDelible对13438名DD先证者进行分析,其中trio模式n=9848,缺少父母一方或双方non-trion=3590。根据上文的过滤条件,从13438个先证者中筛选出260个候选变异。基于人工变异检视,作者认定2/260(0.8%)个变异被错误注释为在基因编码区,17/260(6.5%)个新发变异其实也被父母携带(例如父母假阴性),23/260(8.8%)个变异不太可能是真实存在的(先证者假阳性)。52.2%的假阳性出现在4个先证者中,表明样本选择和/或额外的样本水平QC可进一步降低InDelible的假阳性率。最终有146个SV和大插入可以解释先证者的表型(trio模式56个,non-trio90个)。

有63个以往没有检测到的变异影响已知的DD基因,主要类型是缺失或重复(50/63,79.4%)。其中25个在trio模式下观察到,经过父母验证是新发变异。InDelible在检测21-500bp的变异上尤其有效。此外作者发现最常被影响的基因是MECP2,Rett综合征的致病基因。
作者对这63个变异进行第一轮临床审查,认为9个(14.3%)不能解释先证者表型而排除。之后作者对54个推定致病变异进行了PCR验证,可验证的23个变异100%可确认是真阳性,其中10个是trio模式中的变异,也确认了父母不携带,为新发变异。
作者将54个推定致病变异报告给转诊的临床医师,并由2名高级临床遗传学家进行解释。临床遗传学家将30/54分类为致病或可能致病变异。Non-trio模式发现的变异因为不能确定遗传来源而更难被分类为致病。总而言之,InDelible发现的致病新发变异占整个DD队列的0.7%(18/2592)。
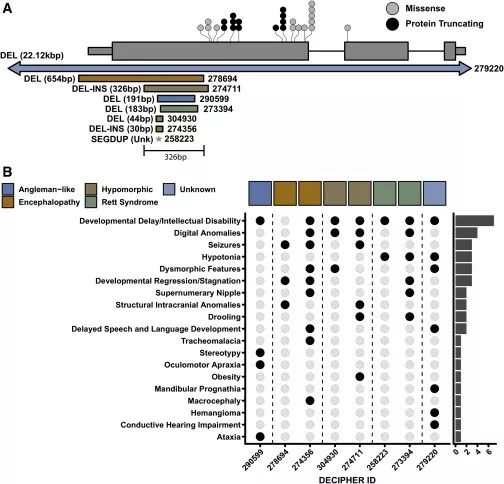
InDelible在MECP2基因中检测到了7个>20bp的新发变异,都预测为截短变异。其中2名先证者的表型为典型的Rett综合征。有趣的是,没有典型Rett综合征表型的5个先证者的SV在326bp区域有交疊。这个低复杂区已经在之前的几项研究中证明高度可变,但是其在MECP2基因中的功能尚不明确。
接下来作者测试了InDelible识别罕见变异的能力。在单等位基因的DD基因中共检测出了145个变异,很大一部分遗传变异是平衡/框内缺失或重复,对目标蛋白的影响不确定(50;34.5%)。其它变异要么主要与非编码序列重叠,要么在具有更可能诊断变异的个体中发现。最后作者向转诊医生报告了7个变异,其中6个被认为是良性的。因此InDelible可有效识别罕见变异,但总体诊断率可能较低。
总结
本文中作者介绍了旨在快速评估ES数据中涉及单基因遗传病的罕见、致病性的隐藏SV断裂点软件InDelible。随后将InDelible应用于DD队列,包含13438个先证者。共检出了146个候选变异,其中63个是未报道过的变异。值得注意的是,作者将长度为21-500bp的变异检出率提高了42.9%。通过对这63个变异的保守临床评估,30个(47.6%)被认为是致病变异,其中最令人感兴趣的是在MECP2中发现的大量截短变异。